

In collaboration with Dr. Diogo Magnani in the Watkins Laboratory in the Dept. of Pathology at the University of Miami’s Miller School of Medicine, we recently received plasma and saliva from two people with Zika virus infections living in the Miami area. Using our amplicon-based approach previously used to sequence Zika virus from travel-related Zika virus cases in Florida, we sequenced the entire coding sequence from the saliva of one individual believed to have been infected in or near Miami. To our knowledge, this represents the first sequenced Zika virus genome from autochthonous mosquito-borne transmission in the United States. The data and analyses can be accessed here.
Preliminary analysis
This sequence (ZL2_Hu0015) is related to other Zika viruses recently sequenced from the Americas (Figure 1), indicating that the current Zika virus outbreak in Florida was the result of an imported infection (human or mosquito) from the ongoing epidemic in the Americas. It is not a result of a separate introduction from Africa or earlier circulating Asian strains.ZL2_Hu0015 is 99.95% identical (5 mismatches) to a Zika virus that we recently sequenced from a traveler coming into Florida from Cuba (ZF10_010U) on June 2nd, 2016. We did not detect any contaminating reads from ZF10_010U in our ZL2_Hu0015 data set (libraries prepared separately with different reagents), demonstrating that these are independent samplings of related viruses. While the data is suggestive, we do not have enough data at this time to conclusively determine the direct origin of the virus. Further sequencing of Zika viruses recovered from autochthonous transmission will help to resolve if there were more than one Zika virus introductions contributing to the outbreak in Florida.
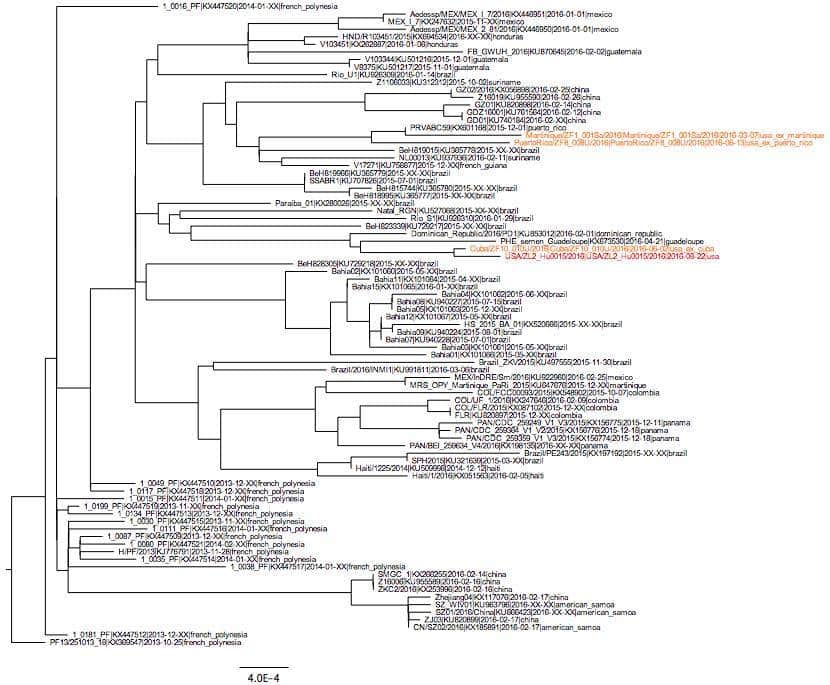
Figure 1. Zika virus tree created using sequences sampled since 2013. Orange indicates sequences from travel-related infections, red indicates the Zika virus sequence from a local infection that occurred in Florida.
We resolved the adaptor-dimer issues that we previously encountered, but we are now working through some issues with reads to our positive control viruses being detected in our samples.
Contamination in sequencing reads
The clinical samples contained very few Zika virus RNA copies (<4 to 38 per μl of RNA), which is a common issue for diagnostic assays and sequencing. As a result, the virus must be amplified for sequencing, which provides opportunities for cross-contamination (aka amplicon “jumping”). We implemented two controls to detect contamination: 1) a no-template water control and 2) we uniquely indexed the multiplexed PCR primer set #1 and #2 amplicons from each sample. We previously detected Zika virus reads in our water control. During the most recent MiSeq run, we detected reads from our positive control Zika virus, PRVABC59, mixed with primer set #2 reads from the clinical samples. For the saliva sample with the most amount of starting Zika virus RNA (~38 per μl of RNA), the contaminating reads were mostly minor compared to the real reads. We used iSNV calling to detect areas with contaminating nucleotides, made assessments to their origin (PRVABC59 or sample), and adjusted the consensus sequence as necessary. Reads aligning to our positive control, however, represented the majority of primer set #2 reads from the samples with very few staring viral RNA copies (<4 to 8 per μl of RNA). We will re-sequence all of these samples using new reagents, however, we are very confident that we are reporting the accurate consensus sequence from patient ZL2.Sa.
Sample
ID = ZL2_Hu0015
Location = Miami, Florida
Date = 2016_08_19
Sample type = Saliva
38 ZIKV copies/μl RNA (21,610 copies/mL saliva)
Data
Our consensus sequences and analyses can be downloaded here. Raw data will be available shortly.
Naming scheme for files
ZL2.c1.SA.a1.l1.r1.S4.L001.R1.[machine generated]
Collection Number = For longitudinal samples, the particular draw the sample was obtained from.
Sample Type = SA = saliva.
Aliquot Number = The aliquot number used for sequences – each sample can have multiple aliquots.
Library Number = The library # for this particular sample. Each sample can have multiple independent libraries (‘biological’ replicate).
Run Number = The # this particular library has run. Each library can be run multiple times (‘technical’ replicate).
Protocol
We based our MiSeq protocol on the protocol developed by Josh Quick and Nick Loman and we are continuously updating it as we make improvements. You can read more here.
Next steps
Together with the Broad Institute, we are currently sequencing three pools of Zika virus-infected adult Aedes aegypti mosquitoes collected in Miami beach. We will also be receiving additional clinical samples from suspected mosquito-borne Zika virus infections in Miami from our collaborators listed below. Data from these samples will provide us with estimates of timing, dynamics, and migration of this just-beginning epidemic in Florida. Data will be made publically available as soon as we are confident in the results.
Collaborators
University of Miami
Diogo Magnani
David Watkins
Paola Lichtenberger
Mike Ricciardi
Varian Bailey
Florida Gulf Coast University
Amanda Tan
Scott Michael
Sharon Isern
Florida Department of Health – Bureau of Public Health Laboratories, Tampa
Marshall Cone
Edgar Kopp
Kelly Hogan
Andrew Cannons
University of Birmingham
Josh Quick
Nick Loman
NextStrain
Trevor Bedford
Richard Neher
Broad Institute
Pardis Sabeti and Crew
Disclaimer
Please note that this data is still based on work in progress and should be considered preliminary. If you intend to include any of these data in publications, please let us know – otherwise please feel free to download and use without restrictions. We have shared this data with the hope that people will download and use it, as well as scrutinize it so we can improve our methods and analyses. Please contact us if you have any questions or comments – we’ll buy beers for #ResearchParasites that spot flaws and faults in the data and come up with improvements! Email us here.







