

Amplicon-based protocol for Zika virus sequencing on the Illumina MiSeq
The protocol can be found here.
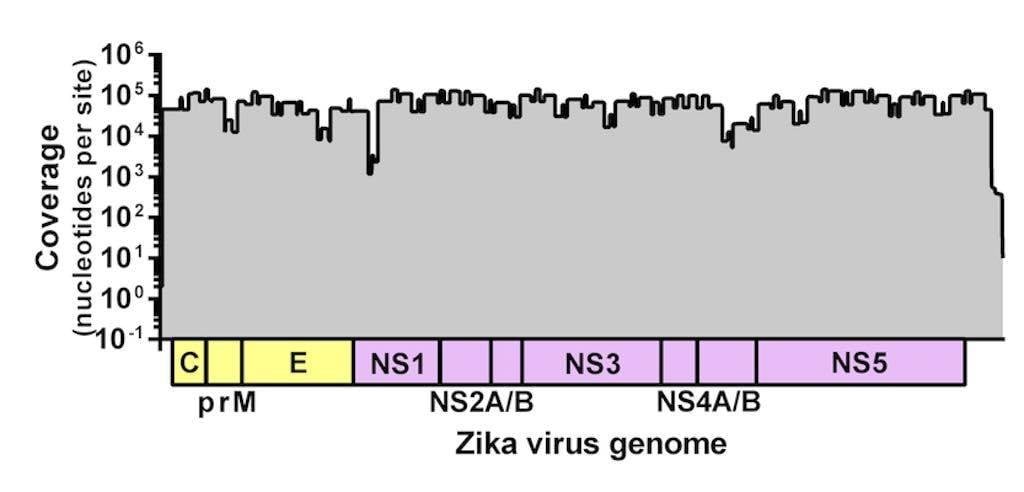
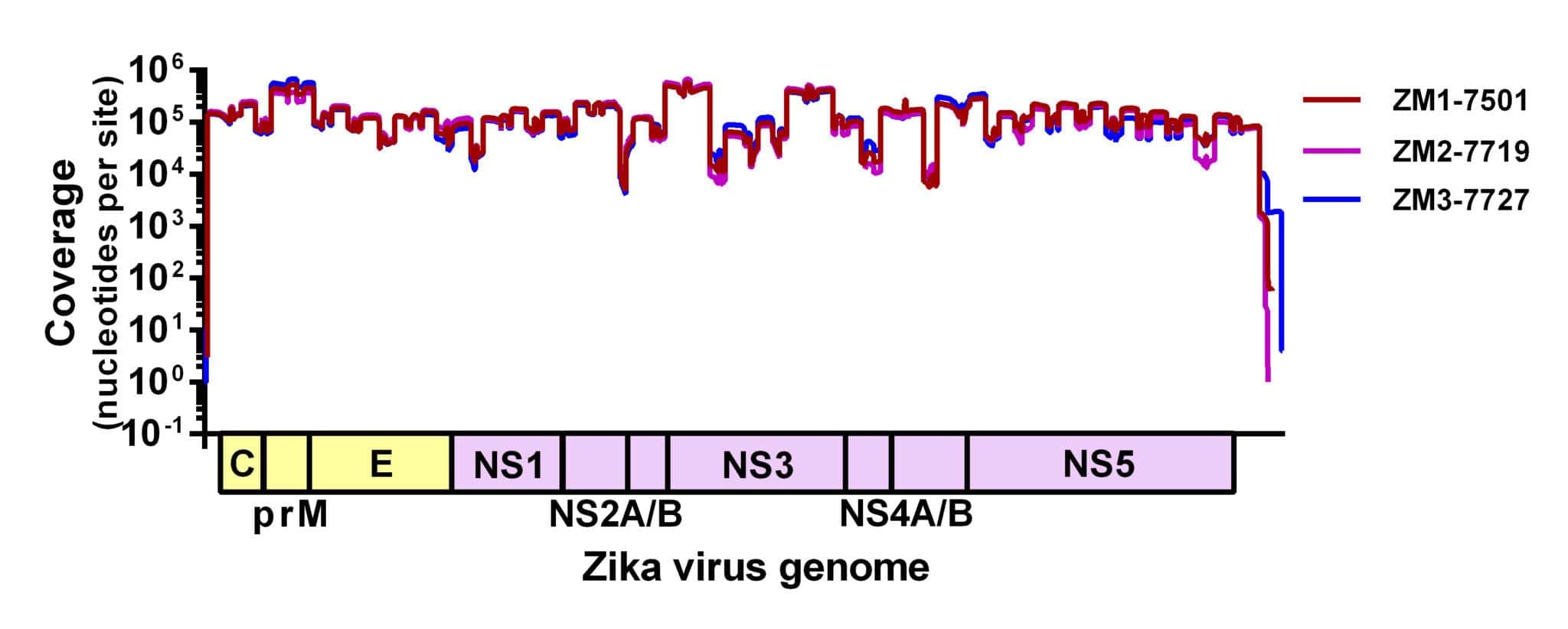
Figure 1. Sequencing coverage of the Zika virus genome. Zika virus strain PRVABC59 spiked with Hela RNA was PCR amplified using 70 virus-specific primers in two reactions, prepared for sequencing using the Kapa Hyper prep kit, and sequenced using the MiSeq v2 500 cycle kit with 7 other libraries. Reads were aligned to the Zika virus genome using NovoAlign. Average coverage depth per site is ~72,000 nt.
Sequencing Zika virus from the ongoing epidemic in the Americas is of critical importance. Data from the viral genome will help us determine how the virus is spreading, changes to viral population sizes, and if the virus is adapting to local conditions. Zika virus, however, does not give up her genetic code so easily. It is not because of any particular challenges with its genome. In fact, we have sequenced a related flavivirus, West Nile virus, with ease from humans, birds, and mosquitoes. The issue is that Zika virus does not produce a very high viremia in humans – meaning there is not much virus to actually sequence. Therefore, unbiased metagenomic “shotgun” approaches reveal limited genetic information about Zika virus (within the high background of host RNA), necessitating more targeted approaches. The question is how to best target the viral genome?
You can target the ~10kb Zika virus genome from two angles, 1) up front via specific primers during PCR to create amplicons spanning the genome, or 2) during library preparation with capture probes. We are exploring both options, but currently only have data from virus-specific amplicons. The amplicon-based approach come in two main flavors: a few long amplicons or many short – both of which have their advantages. Using a protocol from Dave and Shelby O’Connor’s group, we started by amplifying the Zika virus genome in five overlapping ~2 kb segments. The protocol worked great from samples with relatively high virus concentrations. Quickly, however, we discovered that we could not always get amplicons from samples with clinically relevant amounts of virus (i.e., Ct values >33), even after several rounds of optimization. Then Josh Quick and Nick Loman let us try their protocol (developed as part of the ZiBRA project) which includes producing 35 overlapping 400 bp amplicons – in two PCR reactions. I know, we were skeptical too! But we tried it, and behold, we produced solid 400 bp bands from both multiplexed reactions. Then we tried it with decreasing amounts of virus and we still saw bands. From these gel images, however, it was impossible to tell if every primer set produced an amplicon. So we had to sequence it.
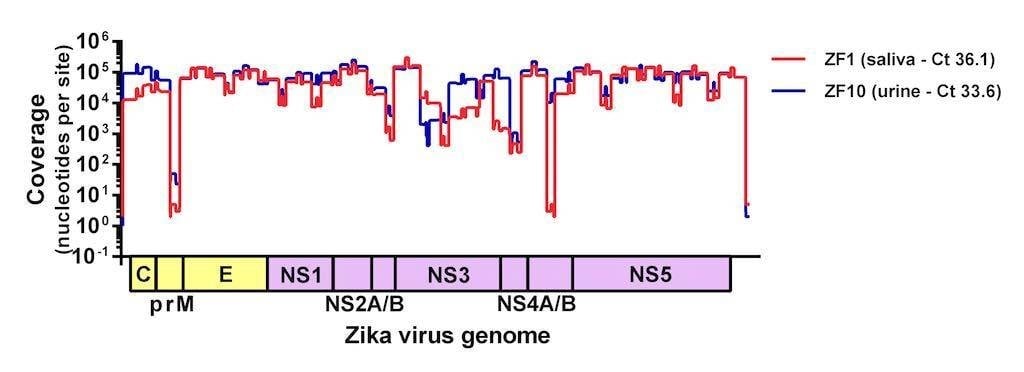
Figure 2. Sequencing coverage of Zika virus from clinical samples. Two samples (saliva and urine) from humans with suspected Zika virus infections were sequenced on the MiSeq using the amplicon-based approach described here. Complete Zika virus coding sequences were recovered from both samples and the protocol is currently being adjusted in an attempt to normalize coverage.
We adapted a simple library prep protocol to sequence the amplicons on our weapon of choice – the MiSeq. We were expecting to find a few gaps in the Zika virus genome in the locations of inefficient amplification. To our astonishment, however, we detected reads spanning the entire coding sequence (importantly, including masking out primer binding sites). Figure 1 shows the coverage plot of a mock sample containing ~100,000 viral copies (strain PRVABC59) and 10ng of Hela RNA amplified using Josh and Nick’s protocol, prepared for sequencing using the Kapa Hyper prep kit, pooled with eight samples, and sequenced using the MiSeq v2 500 cycle kit (2x250nt reads). From this sample, ~3 million reads mapped to the Zika virus genome (~93% of the total), giving us an average coverage depth of ~72,000x.
Since this first run we made a few tweaks to normalize the coverage and streamline the process for MiSeq users. Our version 1 protocol can be found here and will be updated as we make changes. We are also working hard to determine how much virus is required for sequencing (spoiler alert – we have successfully sequenced samples with with Ct >36, Figure 2) and if we can use this approach to accurately call intra-host single nucleotide polymorphisms (iSNVs). This protocol is finally allowing us to sequence Zika virus from clinical samples and to start answering some of the burning questions about the epidemic.
We expect to be able to release our first clinical dataset from Florida shortly.







