Current Data
HLA Typing from Ebola and Lassa Patients
HLA genotype calls from Ebola and Lassa patients admitted in Sierra Leone.
SARS-CoV-1 Evolution
Investigations of the origin and evolution of SARS-CoV-1.
SARS-CoV-2 Evolution
Investigations of the origin and evolution of SARS-CoV-2.
SARS-CoV-2 Genomics
Genome sequencing of SARS-CoV-2 from COVID-19 patients.
West Nile Genomics – WestNile 4K
West Nile virus sequences from across the United States.
Archived Data
Bat Metagenomics
Metagenomic survey of bats from Uganda. The anti-PREDICT Project.
Dengue Florida Genomics
Full-length dengue virus genomes from the 2013 outbreak in Martin County, Florida.
Ebola Epidemiology
Epidemiological data for the 2021 Ebola outbreak in the DRC.
West Nile Genomics – California
West Nile virus sequences from California.
Zika Genomics
Sequence data from the Zika virus outbreak in Florida and beyond – mostly local cases from humans and mosquitoes, but also travel-associated cases.
Zika Epidemiology
We’re compiling Zika case numbers from endemic countries. Our data is based on bar graphs reported and compiled by PAHO.
Zika Functional Evolution
Experimental studies testing Zika virus evolution.
Zika Phylogeography
Phylogeographical analysis of Zika virus spread.
Recent Data-related Blog Posts
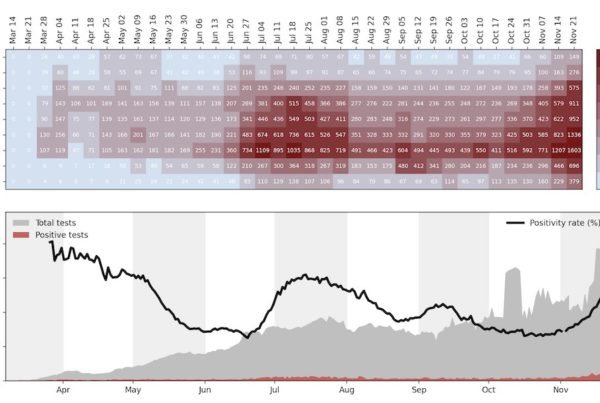
To understand the spread of COVID-19 in San Diego, we looked at the age distribution of COVID-19 over time (Figure 1). Data for this analysis was provided by the Epidemiology and Immunization Services Branch of the County of San Diego Health and Human Services agency through the San Diego Open GIS Data Portal. The visualization was inspired by Gytis Dudas's…
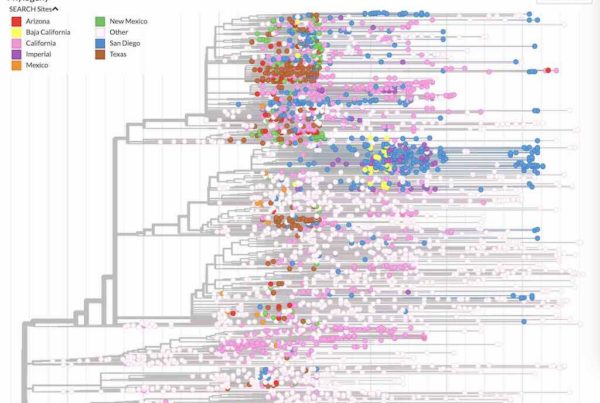
English: To gain insights into the the emergence, spread, and transmission of COVID-19 in our community, we are working with a large number of partners to sequence SARS-CoV-2 samples from patients in San Diego and across the border in Tijuana. In this preliminary analysis we look at the how the outbreaks are connected between California, USA and Baja California, (more…)
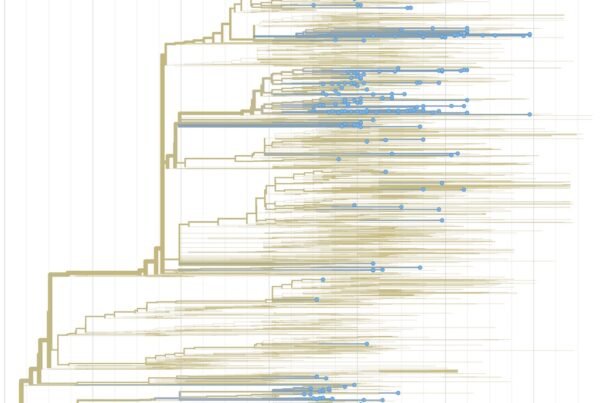
To gain insights into the the emergence, spread, and transmission of COVID-19 in our community, we are working with a large number of partners to sequence SARS-CoV-2 samples from infected patients in San Diego. This is our first preliminary analysis. (more…)